Strand NGS allows alignment of reads to a genome from small RNA, DNA (ChIP-Seq and DNA-Seq applications), and RNA (spliced and unspliced reads) sequencing data generated from platforms of Illumina, Ion Torrent, ABI SOLiD, Roche 454, and Pac Bio. Strand NGS aligner uses a new proprietary algorithm based on the Burrows Wheeler Transform and Smith-Watermann dynamic programming. Other alignment algorithms are often limited to either a specific class of reads or alignment characteristics. However, Strand NGS aligner has been optimized to handle both short reads and long reads, allows an arbitrary number of gaps and mismatches, and handles both single, paired end, and mate paired reads. Strand NGS provides various quality control options to be use pre-alignment or post-alignment of sequencing data..
Download the Data Import, Alignment, and Quality Control Guide White paper on Strand NGS DNA Read Aligner - A Benchmarking study Download the poster on Benchmarking of DNA short read aligners on GCAT data sets Download the Utilities and Biological Contextualization Guide Watch the Alignment with COBWeb Webinar Recording
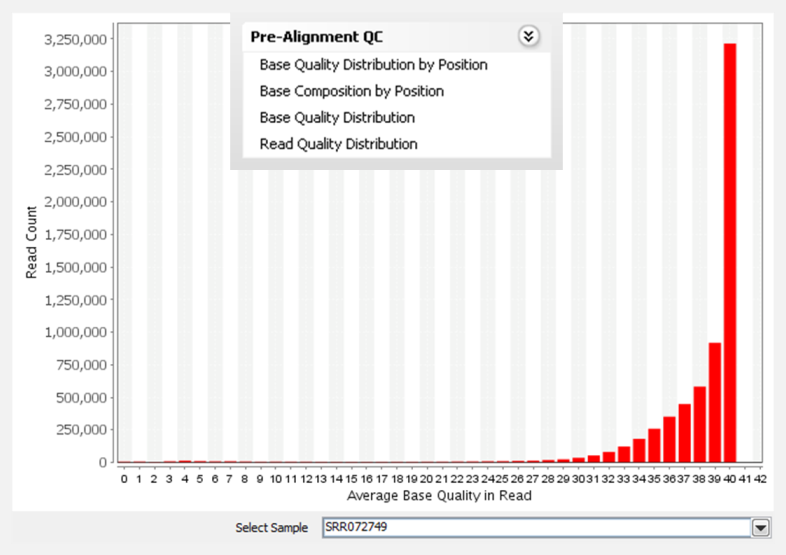
Inspect raw read quality using pre-alignment QC plots, such as base and read level quality distributions, base composition and quality by position.
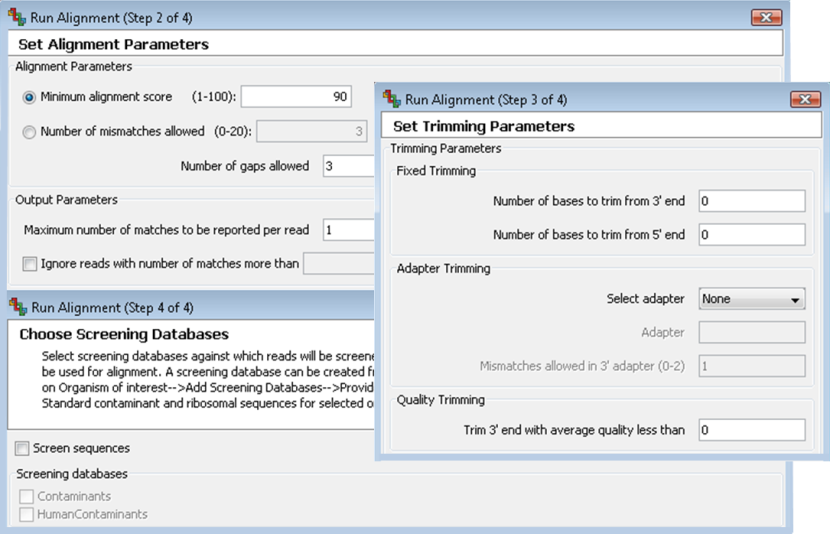
Use options for trimming adaptors and low quality bases, as well as for screening reads against standard screening databases.
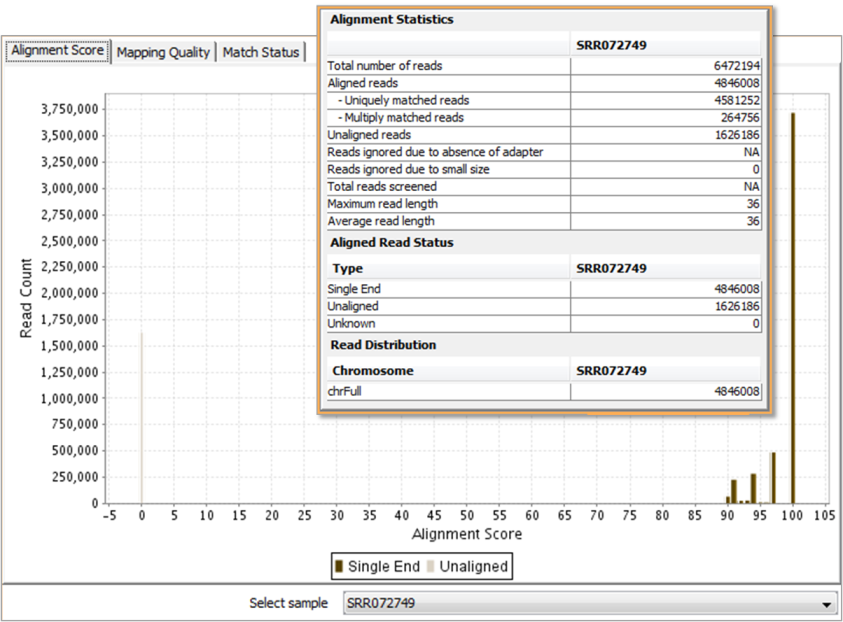
Inspect aligned reads for alignment accuracy and mapping quality. Check and fix mate status in case of paired-end data.
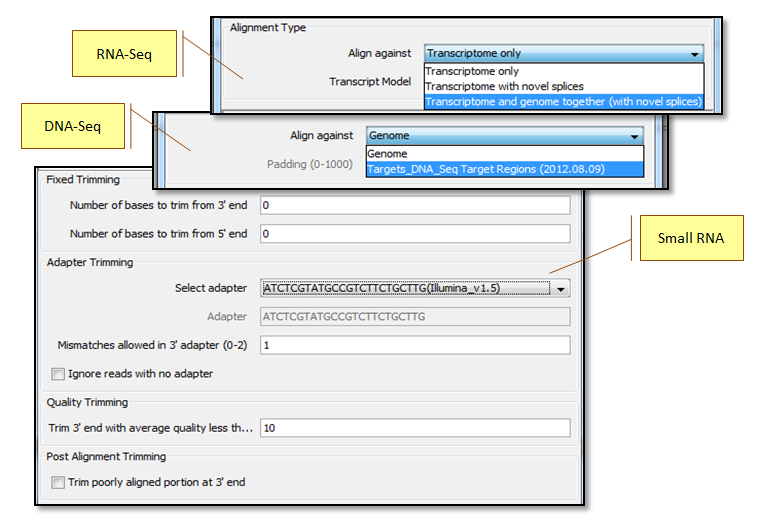
Align against the whole genome or target regions for DNA-Seq; perform split alignment in DNA-Seq for detecting long indels and translocations, align against known transcriptome and/or genome for novel discovery for RNA-Seq; remove adapters for small RNA-Seq.
2018 © Strand Life Sciences Pvt Ltd. All rights reserved.