Strand NGS supports genome-wide methylation analysis using MeDIP-Seq data. The workflow supports data normalization before estimating the methylation signal and identifies differential methylation events across a pair of conditions. These regions can be further annotated based on their location with respect to known genes. Downstream analysis such as GO, pathway analysis can be performed on selected entities.
Download the MeDIP-Seq Highlights Guide Watch the Methyl-Seq Webinar Recording
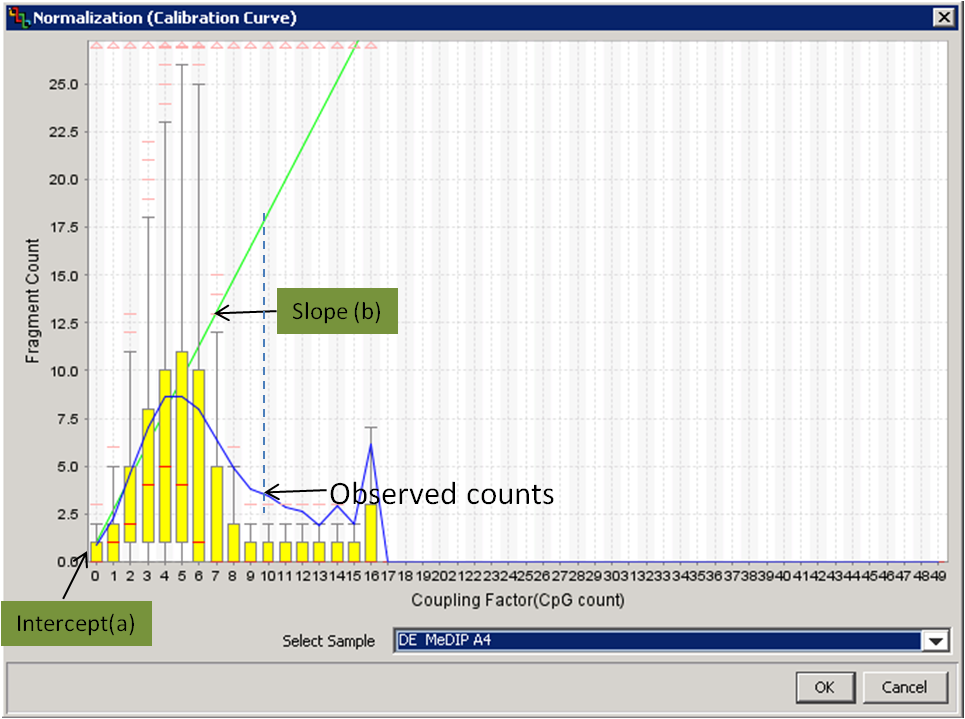
Estimate the linear dependency of methylation signals and CpG counts by calculating the slope and the intercept in a calibration plot.
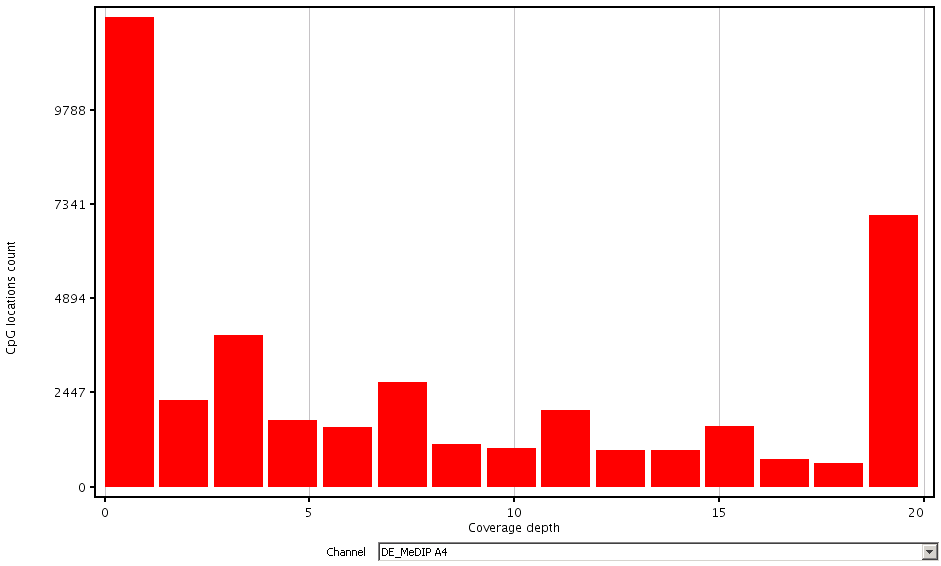
Assess the depth of read coverage across CpG sites.
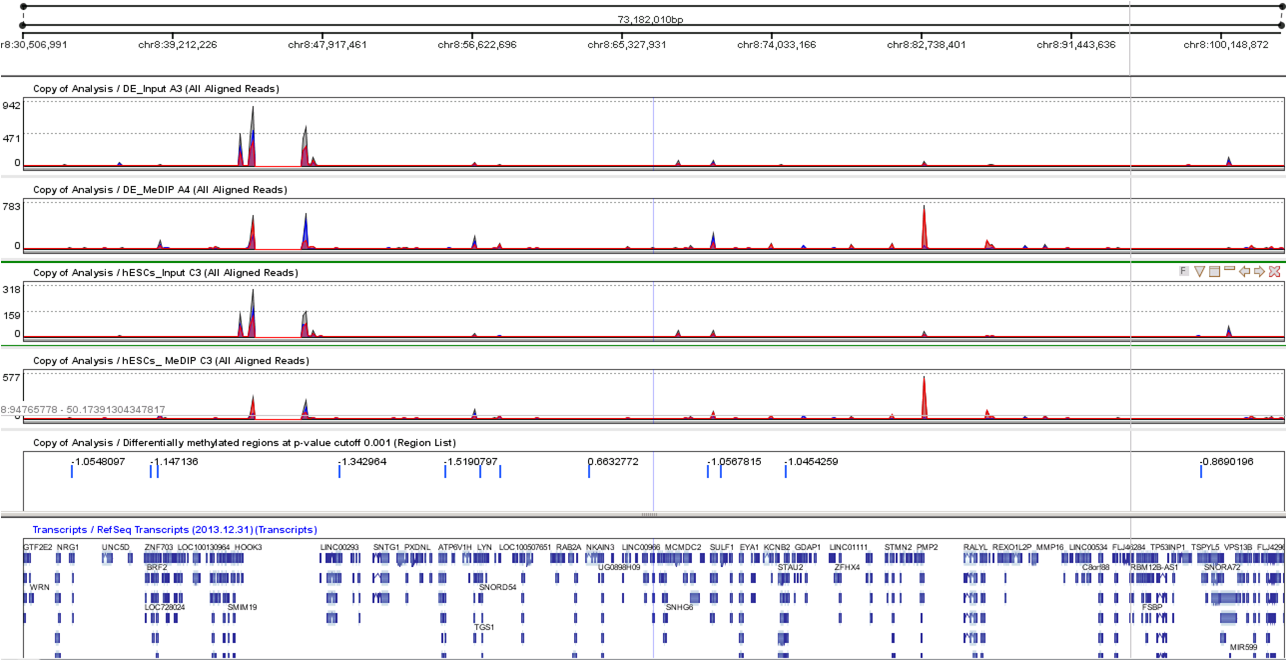
Identify hypomethylated and hypermetylated regions using a p-value threshold. Estimate global and local background noise using input (untreated) samples to help discard false positives.
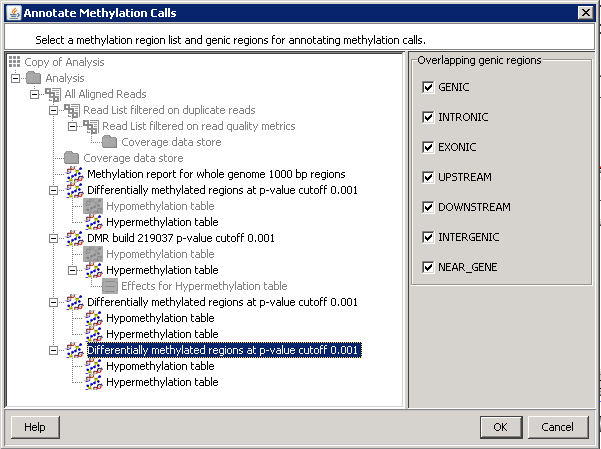
Annotate DMRs for selected overlapping genic regions based on the chosen genes and transcript model.
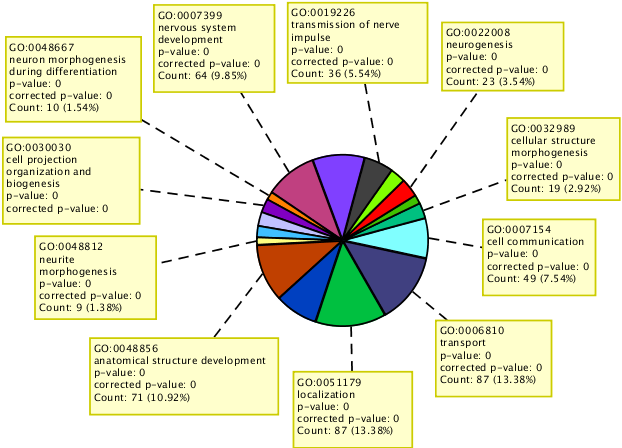
Perform GO analysis on the set of genes for which hypomethylation or hypermethylation is observed in their upstream, 5’ UTR region or exonic region.
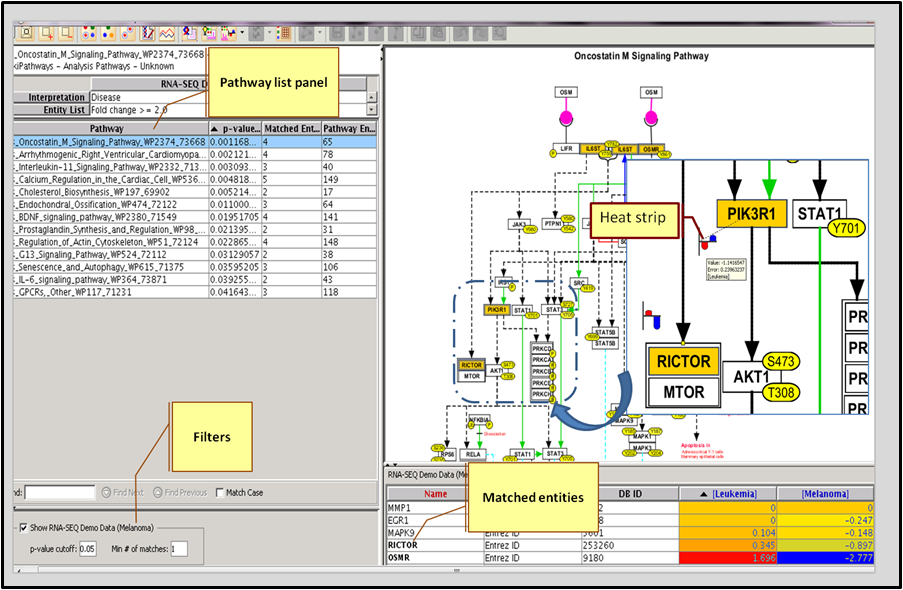
Use the packaged Interaction Database of over 2 million interactions (with supporting PubMed references) or other curated pathways to find relationships between genes. Learn more
2018 © Strand Life Sciences Pvt Ltd. All rights reserved.